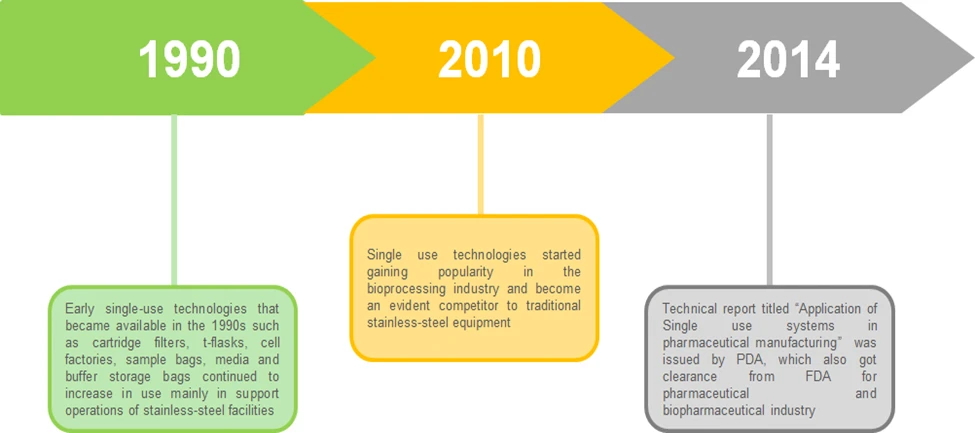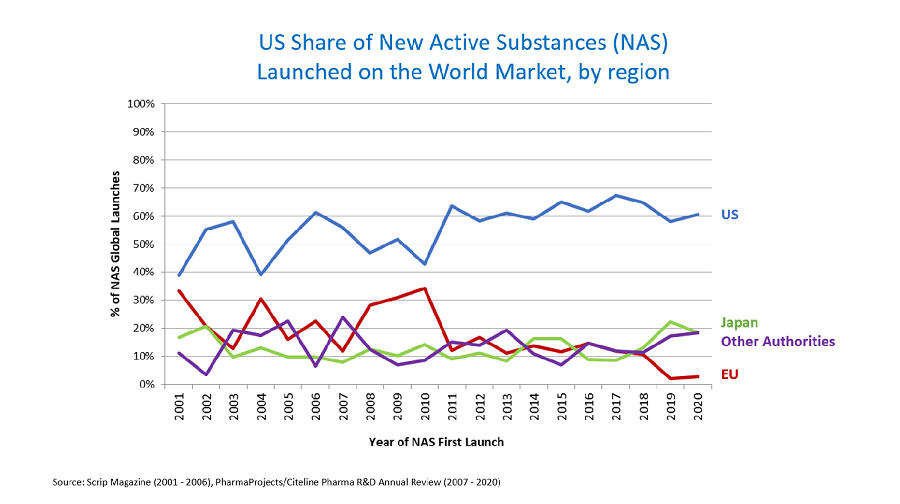
The Prescription Drug User Fee Act (PDUFA), established by Congress in 1992, empowers the FDA to collect fees from pharmaceutical companies that manufacture specific human drugs and biological products. This legislative framework has significantly contributed to streamlining the drug approval process, allowing for more efficient review timelines and resource allocation within the agency.
Since its inception, PDUFA has undergone several amendments, each aimed at refining the fee structure and enhancing the overall efficacy of the drug approval system. These adjustments reflect the evolving landscape of pharmaceutical development and the increasing demand for timely access to innovative therapies. By facilitating a more predictable regulatory pathway, PDUFA not only accelerates market entry for new drugs but also fosters a competitive environment that can drive advancements in drug development.
As the FDA continues to navigate the complexities of modern medicine, the implications of PDUFA are profound for B2B stakeholders across regulatory, QA/QC, CMC, sourcing, and portfolio management sectors. The ongoing dialogue around user fees will be critical in shaping future regulatory strategies and ensuring that the pharmaceutical industry can meet the health needs of a dynamic population.
Start your 7-day trial and see what the database can do →